

Medical device vigilance
The reference regulations on medical devices and active implantable medical devices (MDs) establish that all manufacturers, or their authorised representatives, intending to market their devices in the European Economic Area, should comply with some requirements on the matter of vigilance.
These requirements are clarified in Regulation (EU) 2017/745 as amended (MDR) where manufacturers are requested, not only to report serious incidents to the pertinent Competent Authorities, but also to carry out a trend analysis and report the trends, if any.
Based on the functioning of the incident reporting system, a report can be received by a manufacturer from a lay user, a distributor, a health professional, or even from the competent authority, in case they have been informed by a health professional.
Assessment of reported incidents
When receiving an event, the manufacturer shall carry out an assessment to understand whether this is an incident or a serious incident that, differently from the former, should be reported to the Competent Authority.
If, following such assessment, the event falls into the definition of serious incident (as per art. 2 of the MDR), the manufacturer is obliged to send an initial report to the Competent Authority and to carry out an investigation regarding the reported incident.
This investigation can lead to the need, by the manufacturer, to take a Field Safety Corrective Action (FSCA) that should be disseminated to all users via a Field Safety Notice (FSN).
In this case, the report closing the incident that the manufacturer has to send to Competent Authority at the end of the investigation shall also describe the corrective action taken and copy of the Field Safety Notice to be disseminated to users shall be also included.
In case the investigation results highlight no need to take any corrective action, in the final report submitted to the Competent Authority the manufacturer shall provide an appropriate justification for their decision not to take any corrective action.
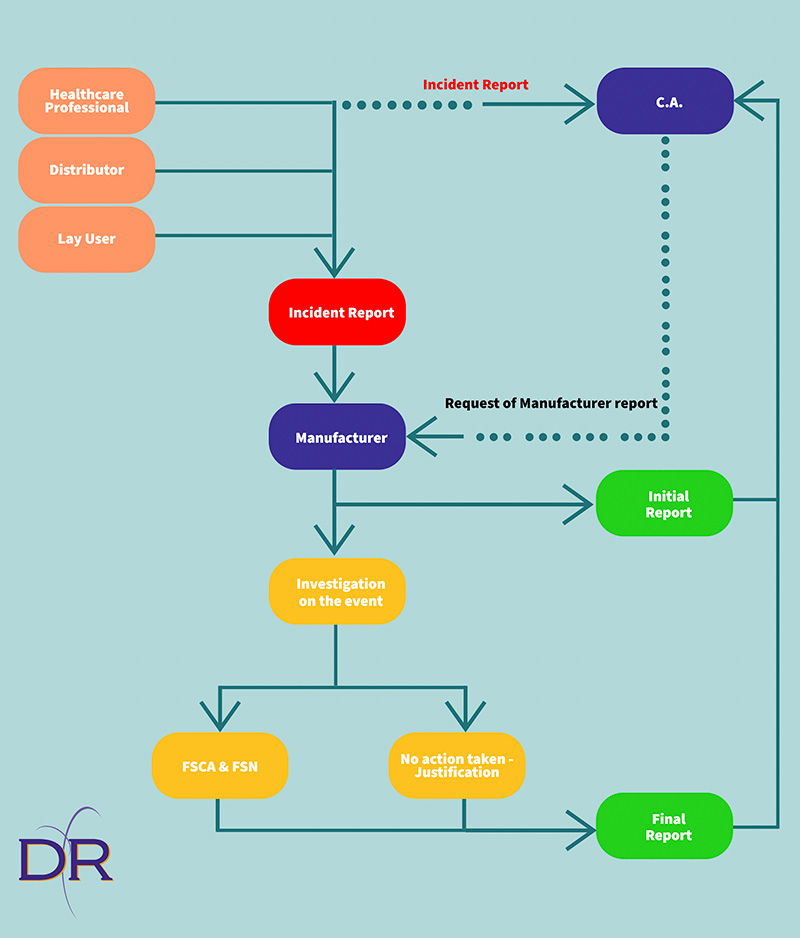
Moreover, as per art. 88 of the MDR, the manufacturer has the obligation to also report
“any statistically significant increase in the frequency or severity of incidents that are not serious incidents or that are expected undesirable side effects that could have a significant impact on the benefit-risk analysis […] The significant increase shall be established in comparison to the foreseeable frequency or severity of such incidents in respect of the device, or category or group of devices, in question during a specific period as specified in the technical documentation and product information.”
Incident reporting timetable
The timetable for the reporting are discussed in art. 87 of Regulation (EU) 2017/745 as amended.
The European guideline MEDDEV 2.12/1, now in its revision no. 8, is essential for the reporting. The guideline provides for the Manufacturer Incident Report (MIR), a form to be used for reports to Competent Authorities until the EUDAMED database becomes fully operative.
Nationally, two memorandum letters have also been released by the Italian Ministry of Health: one dated 8 July 2021 and the other dated 11 October 2022.
They provide operative indications on the methods and timetable for reporting serious incidents, incidents other than serious incidents, complaints, safety corrective actions, as well as the periodic summary reports and the trend reports.
The memorandum letters provide detailed indications to economic operators and users (health professionals, lay users and patients) in terms of what, when and how to report; they apply while waiting for EUDAMED to be fully operative.
Finally, the guideline MDCG 2023-3 – Questions and Answers on vigilance terms and concepts as outlined in the Regulation (EU) 2017/745 on medical devices has the scope of clarifying relevant terms and concepts that are outlined in the MDR in order to obtain an effective and harmonised implementation of the vigilance requirements.
Our device-vigilance consultancy
In the sector of device-vigilance, Di Renzo Regulatory Affairs can assist Clients with several services such as taking on the role of Vigilance Responsible Person either in Italy or in Europe, as well as the role of local contact point in the cases where manufacturers wish to manage vigilance activities themselves.
Moreover, we can take care of the filling in and transmission of notifications, corrective actions and safety notices to the Competent Authorities, and of providing further clarification on request of the Competent Authorities.
We can also assist either manufacturers or their authorised representatives during the investigation regarding an incident contacting, for instance, any reporters for further information, or carrying out specific research in the literature or for safety information in dedicated databases.
We provide our Clients with continuous updating on the evolution of the European and national regulations for this sector and, on request, we can release training sessions for the company’s staff.
We also cooperate with manufacturers to implement, update and manage the post-marketing surveillance and vigilance system, by drafting specific Standard Operating Procedures, collecting feedback information from the market and clinical data, and continuously updating all required technical documents (e.g. post-marketing surveillance plans and reports, PSURs, PMCF plans and reports, clinical assessment and risk management plans and reports).
Written by Ilaria Perretti on 02/16/2023



