

La vigilanza dei dispositivi medici
La normativa di riferimento sui dispositivi medici e dispositivi medici impiantabili attivi (DM) stabilisce che tutti i fabbricanti, o i loro mandatari, che intendono commercializzare i propri dispositivi medici nello Spazio Economico Europeo, devono ottemperare ad alcuni obblighi in tema di vigilanza.
Tali obblighi sono chiariti nel Regolamento (UE) 2017/745 e s.m.i. (MDR) in cui si richiede ai fabbricanti non solo di segnalare gli incidenti gravi alle pertinenti Autorità Competenti, ma anche di eseguire l’analisi dei trend ed eventualmente segnalare le tendenze.
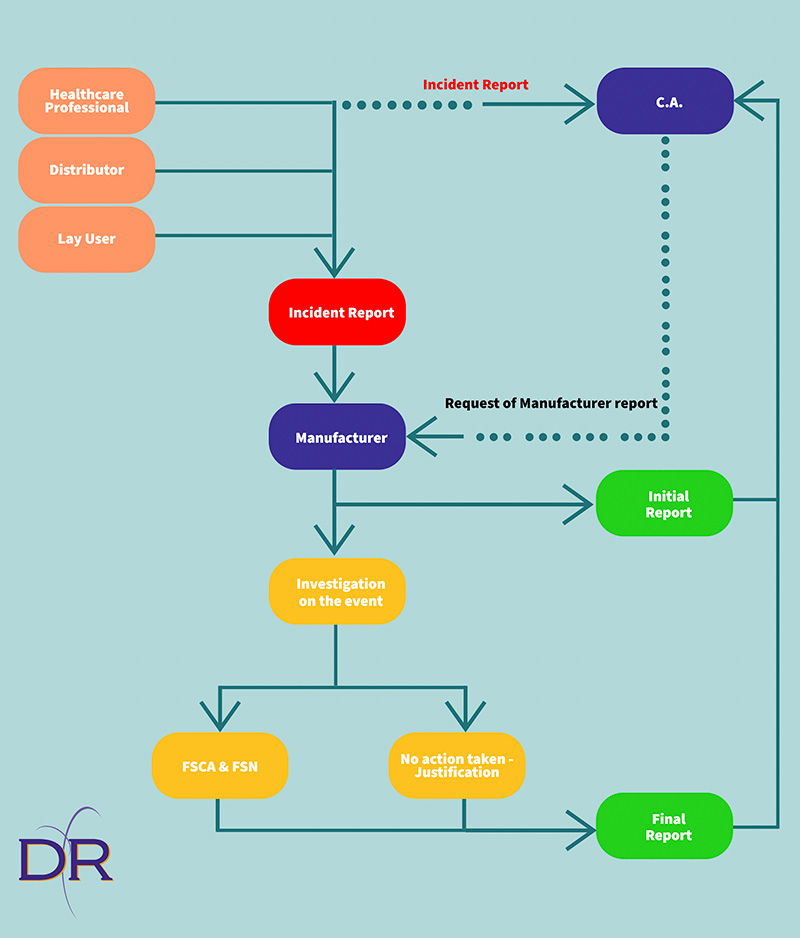
In base al funzionamento del sistema di segnalazione degli incidenti, una segnalazione può pervenire al Fabbricante da un utilizzatore profano, da un distributore, da un operatore sanitario, o anche dall’autorità competente, nel caso in cui sia stata informata da un operatore sanitario.
Valutazione degli incidenti segnalati
Nel momento in cui riceve un evento, il Fabbricante deve svolgere una valutazione per capire se si tratta di un incidente o di un incidente grave, che a differenza del primo deve essere segnalato all’Autorità Competente.
Se, a seguito di valutazione, l’evento ricade nella definizione di incidente grave (come da art. 2 del MDR), il Fabbricante ha l’obbligo di inviare un report iniziale all’Autorità Competente e di svolgere un’indagine relativamente all’evento segnalato.
L’indagine può portare alla necessità, da parte del Fabbricante, di intraprendere una Azione correttiva in Campo (FSCA), che deve essere divulgata a tutti gli utilizzatori tramite un Avviso di Sicurezza (FSN).
In questo caso, nel report di chiusura dell’incidente che il Fabbricante deve inviare all’autorità Competente a conclusione dell’indagine, deve essere descritta anche l’azione correttiva intrapresa e deve essere inviata copia dell’avviso di sicurezza che sarà divulgato agli utilizzatori.
Nel caso in cui i risultati dell’indagine non evidenzino la necessità di intraprendere alcuna azione correttiva, nel report finale inviato all’Autorità competente il Fabbricante deve fornire una opportuna giustificazione per la decisione di non intraprendere alcuna azione correttiva.
Inoltre come da art. 88 del MDR, il fabbricante ha l’obbligo di segnalare anche:
“ogni aumento statisticamente significativo della frequenza o della gravità di incidenti che sono diversi da quelli gravi o effetti collaterali indesiderati attesi che possano avere un impatto significativo sull’analisi dei rischi e dei benefici.[…] L’aumento significativo è stabilito in rapporto alla frequenza o alla gravità prevista di tali incidenti, relativi al dispositivo o alla categoria o al gruppo di dispositivi in questione, in un periodo determinato precisato nella documentazione tecnica e nelle informazioni sul prodotto”.
Le tempistiche delle segnalazioni di incidenti
Le tempistiche di segnalazione sono discusse all’interno dell’art. 87 del Regolamento (UE) 2017/745 e s.m.i.
Ai fini delle modalità di segnalazione, importante è la linea guida europea MEDDEV 2.12/1, giunta alla sua ottava revisione, nella quale è fornito il Manufacturer Incident Report (MIR), il modello da utilizzare per le segnalazioni alle Autorità Competenti fino a quando la banca dati EUDAMED non sarà pienamente operativa.
Anche a livello nazionale sono state rilasciate due circolari dal Ministero della Salute, una dell’8 luglio 2021 e una dell’11 ottobre 2022 che forniscono indicazioni operative sulle modalità e le tempistiche delle segnalazioni di incidenti gravi, incidenti diversi da quelli gravi, dei reclami, delle azioni correttive di sicurezza, nonché delle relazioni di sintesi periodiche e delle relazioni sulle tendenze.
Le circolari danno indicazioni dettagliate agli operatori economici e agli utilizzatori (operatori sanitari, utilizzatori profani e pazienti) in termini di cosa, quando e come segnalare; esse valgono in attesa della piena operatività di EUDAMED.
Infine, la linea guida MDCG 2023-3 – Questions and Answers on vigilance terms and concepts as outlined in the Regulation (EU) 2017/745 on medical devices ha lo scopo di chiarire termini e concetti importanti delineati nel MDR al fine di ottenere un’attuazione efficace e armonizzata dei requisiti di vigilanza.
La nostra consulenza di vigilanza dei dispositivi medici
Di Renzo Regulatory Affairs nel settore della device-vigilanza è in grado di assistere i propri Clienti con vari servizi come l’assunzione del ruolo di Responsabile per la vigilanza in Italia o in Europa, su mandato del Fabbricante, o anche il ruolo di contact point locale, nei casi in cui un Fabbricante voglia mantenere presso di sé la gestione delle attività di vigilanza.
Inoltre possiamo occuparci della compilazione e trasmissione alle Autorità Competenti delle notifiche di incidenti, azioni correttive e avvisi di sicurezza, e di fornire ulteriori chiarimenti su richiesta delle Autorità Competenti.
Possiamo anche assistere i Fabbricanti o i loro Mandatari nella fase di indagine relativa ad un incidente, ad esempio contattando i segnalatori per ulteriori informazioni, o svolgendo specifiche ricerche di letteratura o di informazioni di sicurezza su database dedicati.
Ai nostri Clienti forniamo aggiornamenti continui sull’evoluzione della normativa di settore Europea e nazionale, e possiamo, su richiesta, erogare delle sessioni di formazione per personale aziendale.
Collaboriamo inoltre con i Fabbricanti anche per implementare, aggiornare e gestire il sistema di sorveglianza post-marketing e vigilanza, attraverso la redazione di specifiche procedure operative Standard, la raccolta di informazioni di ritorno dal mercato, di dati clinici e l’aggiornamento continuo di tutta la documentazione tecnica necessaria (come piani e report si sorveglianza post-commercializzazione, PSUR, piani e report di PMCF, piani e report di valutazione clinica e di gestione del rischio).
Scritto da Ilaria Perretti il 16/02/2022


